Come il Regno Unito ha fatto prima sul vaccino contro il coronavirus
E perché l'autorizzazione arrivata in anticipo rispetto a Stati Uniti e Unione Europea ha fatto discutere e ha portato a qualche critica
Mercoledì 2 dicembre il Regno Unito è diventato il primo paese ad autorizzare l’utilizzo su larga scala del vaccino contro il coronavirus sviluppato da Pfizer-BioNTech. La decisione consentirà di avviare la distribuzione delle prime dosi già a iniziare dalla prossima settimana, ma ha fatto sollevare alcune perplessità e critiche, sia nei confronti del governo britannico per avere accelerato un’autorizzazione così delicata, sia nei confronti dell’Unione Europea accusata da alcuni esponenti politici di essere in ritardo con l’approvazione.
Il governo britannico confida che il vaccino possa contribuire a rallentare la pandemia da coronavirus, alleviando la crisi sanitaria nel paese che ha finora causato la morte di circa 60mila persone. Lo confidano anche altri paesi, compresi gli Stati Uniti, dove sono in corso le verifiche sui dati forniti da Pfizer-BioNTech, un processo lungo e che in condizioni normali richiede mesi per essere completato. Le stesse procedure sono in corso per il vaccino di Moderna, e saranno utilizzate per gli altri vaccini sperimentali non appena avranno terminato la parte più rilevante dei loro test clinici.
Per capire come il Regno Unito sia arrivato prima degli altri, occorre fare qualche passo indietro.
Autorizzazione di emergenza
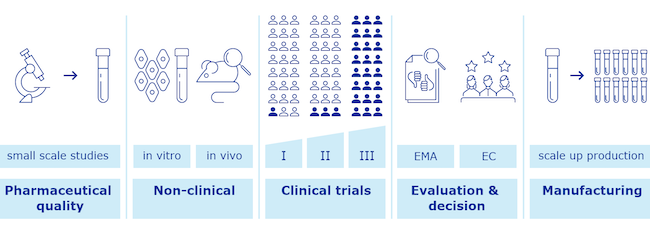
In linea generale, prima di essere impiegato sulla popolazione, un farmaco deve ricevere una serie di autorizzazioni da parte delle autorità di controllo. Le due più grandi e importanti in Occidente sono la Food and Drug Administration (FDA) degli Stati Uniti e l’Agenzia europea per i medicinali (EMA) dell’Unione Europea. Le due agenzie hanno caratteristiche simili, con la prima che ha una gestione maggiormente centralizzata.
Nel caso di particolari circostanze, come la pandemia da coronavirus in corso, le due agenzie prevedono che l’approvazione di farmaci e vaccini possa seguire un processo di verifica accelerato, che porta a un’autorizzazione di emergenza. Ciò consente di stringere i tempi delle verifiche e di offrire prima terapie e vaccini che potrebbero contribuire a salvare vite, alleviare il carico di lavoro per i sistemi sanitari e nel caso di un’epidemia ridurre la circolazione dell’agente che la causa.
Per il processo di autorizzazione di emergenza sono previsti protocolli e procedure per bilanciare rischi e benefici: approvare più velocemente un farmaco/vaccino implica l’assunzione di qualche minimo rischio in più, renderlo disponibile il prima possibile può portare benefici a milioni di persone. Di solito, più i dati forniti dalle aziende farmaceutiche che richiedono l’approvazione sono accurati, coerenti e ben strutturati, più è probabile che sia concessa l’autorizzazione di emergenza.
Una volta che un farmaco/vaccino è stato autorizzato con un percorso accelerato, gli organismi di controllo proseguono nelle loro verifiche e raccolgono inoltre i dati sull’impiego del trattamento sulla popolazione. Ciò consente di intervenire rapidamente per sospendere l’autorizzazione, nel caso in cui qualcosa vada storto. L’obiettivo è accorgersene basandosi su dati e indicatori, prima che ci possano essere eventuali rischi per la popolazione.
Efficacia e sicurezza
Il vaccino di Pfizer-BioNTech ha superato con risultati promettenti le tre fasi di sperimentazione clinica, orientate a verificare inizialmente la sicurezza della soluzione e solo in un secondo momento la sua efficacia. Nella fase 3 della sperimentazione sono stati coinvolti 44mila volontari, con un’incidenza relativamente bassa di effetti avversi, di lieve entità e temporanei come mal di testa o dolore nel punto dell’iniezione. L’efficacia rilevata è stata del 95 per cento, molto al di sopra delle aspettative. Il vaccino di Moderna ha fatto rilevare un’efficacia del 94,1 per cento e non ha fatto riscontrare particolari effetti avversi.
Richiesta di autorizzazione
Pfizer-BioNTech e Moderna negli ultimi giorni hanno presentato alla FDA e all’EMA la documentazione sui test clinici effettuati, chiedendo un’autorizzazione di emergenza per i loro rispettivi vaccini.
L’FDA ha annunciato che il vaccino di Pfizer-BioNTech sarà rivisto da una prima commissione di esperti il prossimo 10 dicembre, mentre per quello di Moderna dovrebbe esserci una revisione il 17 dicembre. La commissione valuta i dati dei test clinici e poi raccomanda o meno l’impiego del vaccino, che viene deciso in ultima istanza dall’FDA.
Nell’Unione Europea la responsabilità di rivedere i dati sui test clinici spetta all’EMA, che fornisce poi una raccomandazione sull’impiego o meno del vaccino alla Commissione Europea, che ha il compito di formalizzare la scelta. L’EMA ha chiarito che svolgerà una revisione al più tardi il prossimo 29 dicembre, ma è possibile che il tutto si svolga prima.
L’annuncio della data è stato ripreso strumentalmente da alcuni esponenti politici, come il senatore Matteo Salvini della Lega, che ha accusato l’EMA di aspettare fino al 29 dicembre per prendere una decisione, omettendo che il riferimento temporale indicato dall’agenzia comprendesse un “al più tardi”.
In questi mesi, sia l’FDA sia l’EMA hanno comunque lavorato sui dati che Pfizer-BioNTech e Moderna fornivano man mano che progredivano i loro test clinici. L’EMA, per esempio, ha avviato questa revisione “in corsa” il 6 ottobre scorso, proprio per raccogliere e analizzare le informazioni il più in fretta possibile sul vaccino di Pfizer-BioNTech.
Tenendo a mente tutto questo, ora arriviamo al Regno Unito.
Più veloci
Dallo scorso 30 gennaio il Regno Unito ha formalmente lasciato l’Unione Europea per via di Brexit, ed è entrato in un periodo di transizione che dovrebbe terminare con la fine di quest’anno. Il cambiamento riguarda anche l’EMA: il Regno Unito ne fa ancora parte, ma dal primo febbraio non ha alcun ruolo sulle sue attività. In pratica, le leggi europee sui farmaci si applicano ancora nel Regno Unito, ma questo non partecipa ai comitati scientifici, alle commissioni e all’amministrazione dell’EMA.
Il Regno Unito avrebbe quindi potuto attendere la decisione dell’EMA sul vaccino, come hanno deciso di fare gli altri stati membri dell’Unione Europea, ma ha invece preferito seguire una propria strada. La sua agenzia nazionale per il controllo dei farmaci, MHRA (Medicines and Healthcare products Regolatory Agency), lo scorso 30 ottobre ha avviato una propria valutazione in corsa dei dati che Pfizer-BioNTech rendevano via via disponibili agli organismi di controllo sui loro test clinici.
L’MHRA ha poi ritenuto che quei dati fossero sufficienti per concedere l’autorizzazione, anche prima di un’analisi più approfondita sui risultati finali forniti da Pfizer-BioNTech al momento della formalizzazione della propria richiesta per l’approvazione. Per procedere indipendentemente dall’EMA, il Regno Unito ha sfruttato un’eccezione sui processi di autorizzazione prevista in una direttiva europea del 2001. La norma prevede che uno stato membro possa autorizzare, in via temporanea, la distribuzione di un farmaco/vaccino non ancora approvato “per affrontare la diffusione sospetta o confermata di agenti patogeni, tossine, agenti chimici o radiazioni nucleari che potrebbero causare danno” alla popolazione.
La pandemia da coronavirus in corso risponde a queste caratteristiche e ha quindi indotto il Regno Unito ad agire in autonomia. La decisione ha fatto sollevare perplessità e qualche critica, considerato che il processo di approvazione è ancora in corso presso le due più grandi e importanti autorità di controllo per i farmaci in Occidente.
La responsabile dell’MHRA, June Raine, ha però difeso la scelta della propria agenzia: “Non sono state prese scorciatoie”. Raine ha dato risposte un poco più evasive quando le è stato chiesto in cosa fosse diverso l’approccio britannico rispetto a quello dell’EMA o della FDA, sostenendo che il processo seguito sia stato “equivalente a quello di tutti gli altri standard internazionali”. L’autorizzazione di emergenza consentirà di distribuire le prime dosi, e intanto l’MHRA lavorerà per fornire la piena approvazione attraverso i tempi che normalmente queste attività richiedono.
27
I 27 stati membri dell’Unione Europea per ora non hanno dato indicazioni su possibili accelerazioni nel processo decisionale. L’orientamento è di arrivare uniti e nei tempi necessari, sia per garantire la sicurezza della popolazione sia per non fornire messaggi contrastanti.
Se si riveleranno efficaci come indicato dai test clinici, i vaccini potranno essere uno strumento importante per rallentare la pandemia e tornare alla normalità, e per questo il processo con cui saranno approvati dovrà guadagnarsi la fiducia di milioni di persone. In mancanza di questa, le campagne vaccinali potrebbero rilevare una bassa partecipazione, riducendo il contenimento della pandemia.
La responsabile dell’EMA, Emer Cooke, ha rassicurato i ministri della Salute degli stati membri nel corso di un loro incontro informale sull’attività della sua agenzia. Ha spiegato che l’autorizzazione di emergenza sarà concessa sulla base delle evidenze scientifiche, con dati sufficienti per ritenere che i benefici superino rischi eventuali.
Tempo
I tempi dell’autorizzazione di emergenza del vaccino di Pfizer-BioNTech da parte dell’EMA non sono ancora chiari, per questo per ora c’è solamente la data indicativa e di massima del 29 dicembre “al più tardi”. Oltre ai tempi dell’analisi ce ne saranno altri di natura tecnica: l’EMA dovrà fornire la raccomandazione alla Commissione Europea, che dovrà poi preparare i documenti condividendo la propria decisione con gli stati membri.
Ci saranno poi tempi tecnici per avviare la distribuzione delle prime dosi, come ha spiegato ieri il ministro della Salute, Roberto Speranza. Il governo italiano prevede di avviare la distribuzione gratuita delle prime dosi, agli individui a rischio, verso la fine del prossimo gennaio.
Entro il 12 gennaio l’EMA valuterà anche il vaccino di Moderna, le cui dosi potrebbero essere distribuite a partire da febbraio, migliorando l’offerta rispetto all’alta domanda di vaccini. La Commissione Europea in questi mesi ha prenotato centinaia di milioni di dosi dai principali produttori, anche se alcune delle loro soluzioni sono ancora nella fase sperimentale.
Cautele
L’approvazione dei vaccini di Pfizer-BioNTech e Moderna consentirà di avere un nuovo strumento per arginare la pandemia da coronavirus. Mentre è incoraggiante che in appena dieci mesi due vaccini siano passati dal laboratorio ai test clinici e infine alle autorità di controllo, è bene ricordare che non abbiamo ancora tutte le risposte sul ruolo che queste soluzioni potranno avere nel risolvere l’attuale crisi.
Solo dopo l’impiego su milioni di persone potremo sapere quale sia l’efficacia nella comunità del vaccino, rispetto a quella rilevata nei test clinici con un numero limitato di individui (seppure piuttosto ampio). I due vaccini, come quelli ora in sperimentazione, esistono da poco tempo e non è quindi possibile stabilire quale grado di protezione offrano, soprattutto per valutare se portino a un’immunità nel lungo periodo.



