Questa intelligenza artificiale risolve un grande problema
Da sempre i ricercatori faticano a ricostruire forma e funzione delle proteine: AlphaFold lo fa in pochissimo tempo
di Emanuele Menietti
Un’intelligenza artificiale (AI) della divisione DeepMind di Google è riuscita a risolvere in poco tempo uno dei più grandi problemi della biologia: determinare la forma di una proteina, partendo dalle catene di amminoacidi che la costituiscono. In alcuni casi, l’AI ha offerto un risultato con un alto grado di affidabilità dopo appena 30 minuti di calcolo, a fronte degli anni – talvolta decenni – richiesti per ottenere risultati simili con i metodi sperimentali. Secondo diversi esperti, il nuovo sistema potrebbe rivoluzionare parte della ricerca, accelerando la scoperta di nuovi farmaci e trattamenti.
Forma e funzione
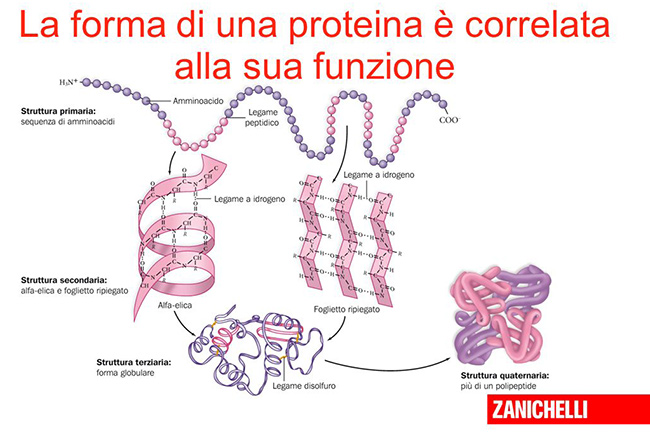
Le proteine sono centrali nella nostra esistenza e in quella degli altri esseri viventi: regolano il metabolismo, la risposta agli stimoli e sono essenziali per il trasporto delle molecole, solo per fare qualche esempio. I vari tipi di proteine hanno proprie caratteristiche determinate dalle catene di amminoacidi che le costituiscono e dalla forma che assumono, ripiegandosi su loro stesse. La forma è essenziale nel determinare la funzione, e per questo in biologia molecolare si dice spesso che “la struttura è la funzione” di una proteina.
La maggior parte delle proteine ha dimensioni comprese tra 1 e 100 nanometri (un nanometro equivale a un miliardesimo di metro) ed è quindi molto difficile studiarne la struttura. Negli ultimi decenni, buona parte degli esperimenti di biologia molecolare ha riguardato proprio le tecniche e i metodi da impiegare per provare a comprendere come specifiche proteine si ripieghino su loro stesse.
Grazie a una tecnica che sfrutta i raggi X (cristallografia a raggi X), a partire dagli anni Cinquanta divenne possibile determinare la struttura di alcune proteine. Le tecniche si sarebbero poi affinate ulteriormente nell’ultimo decennio grazie alla microscopia crioelettronica, che prevede l’osservazione dei campioni a temperature estremamente basse, offrendo migliori risultati e riducendo il rischio di modificare le molecole nella fase di preparazione del materiale da osservare.
Previsione al computer
Anche se ha permesso negli anni di osservare la struttura di diverse proteine, rendendo possibile lo studio del modo in cui interagiscono negli organismi, l’approccio sperimentale richiede molto tempo per essere messo in pratica e porta spesso a risultati poco soddisfacenti. Comprendere l’effettiva forma tridimensionale della proteina dalle osservazioni al microscopio nell’infinitamente piccolo, dove i campioni appaiono come se fossero bidimensionali, non è semplice e richiede spesso anni di lavoro, senza garanzie di arrivare a qualcosa di concreto.
Per questo motivo, già a partire dagli anni Ottanta, alcuni ricercatori si chiesero se non fosse possibile seguire un approccio diverso: partire dalle catene di amminoacidi e scoprire come queste determinino la struttura tridimensionale delle proteine di cui fanno parte (negli amminoacidi non ci sono istruzioni su che forma debba assumere la proteina: i ripiegamenti sono dovuti alle leggi della fisica). I ricercatori pensarono di sfruttare una risorsa che 40 anni fa iniziava a dimostrare di avere crescenti capacità di calcolo: i computer.
I primi modelli informatici si rivelarono però poco affidabili, soprattutto dal punto di vista della riproducibilità dei risultati, uno dei pilastri del metodo scientifico. Un modello che si era rivelato adeguato nel determinare la struttura di una proteina, falliva miseramente se applicato da altri ricercatori per i loro studi su proteine di altro tipo.
Competizione
Desideroso di migliorare le cose, a metà anni Novanta il biologo computazionale John Moult (Università del Maryland, Stati Uniti) fondò insieme ad altri ricercatori il Critical Assessment of Structure Prediction (CASP), un’iniziativa per mettere in competizione ogni due anni i centri di ricerca stimolandoli a produrre nuove soluzioni al computer per la previsione delle strutture delle proteine.
La sfida consiste nel prevedere una struttura proteica già nota attraverso i classici metodi sperimentali, ma non ancora resa pubblica. Secondo gli organizzatori, in circa 35 anni di esistenza, CASP ha permesso non solo di incentivare la ricerca nel settore, ma anche di fare selezione, scartando gli approcci meno promettenti.
AlphaFold
Nel 2018, DeepMind aveva partecipato al CASP13 con AlphaFold, un modello che aveva dato alcuni risultati incoraggianti, con tecniche comunque piuttosto comuni di intelligenza artificiale e già impiegate da altri concorrenti. Semplificando molto, il sistema prevedeva di utilizzare tecniche di apprendimento automatico (deep learning) basandosi su una grande mole di dati per prevedere la distanza tra le coppie di amminoacidi nelle catene che costituiscono le proteine. AlphaFold utilizzava poi le informazioni derivate dall’analisi automatica di quei dati per modellare la proteina, effettuando autonomamente una verifica sulla propria previsione.
Era un approccio piuttosto ortodosso, ma non portava a risultati convincenti a sufficienza. I ricercatori di AlphaFold decisero quindi di provare una strada diversa, dando maggior peso alle informazioni note sui meccanismi fisici e geometrici che limitano il modo in cui le proteine possono ripiegarsi, assumendo poi la loro forma finale. Decisero inoltre di sviluppare la loro intelligenza artificiale per orientarla alla previsione della forma di singole porzioni delle proteine, dando minore rilevanza alla previsione delle relazioni tra gli amminoacidi.
Al CASP14 organizzato quest’anno, a distanza a causa della pandemia da coronavirus, AlphaFold nella sua nuova versione ha sorpreso i giudici della competizione e numerosi altri esperti di biologia molecolare. In circa i due terzi dei casi, l’intelligenza artificiale ha dato risultati sulla forma delle proteine comparabili a quelli ottenuti con le osservazioni sperimentali.
Il modello ha ottenuto un punteggio di 90, su un massimo di 100 che indica la totale corrispondenza alla struttura basata sull’osservazione sperimentale della forma della proteina. Un punteggio simile è considerato più o meno equivalente a quello ottenibile con l’osservazione diretta, dicono i responsabili di CASP.
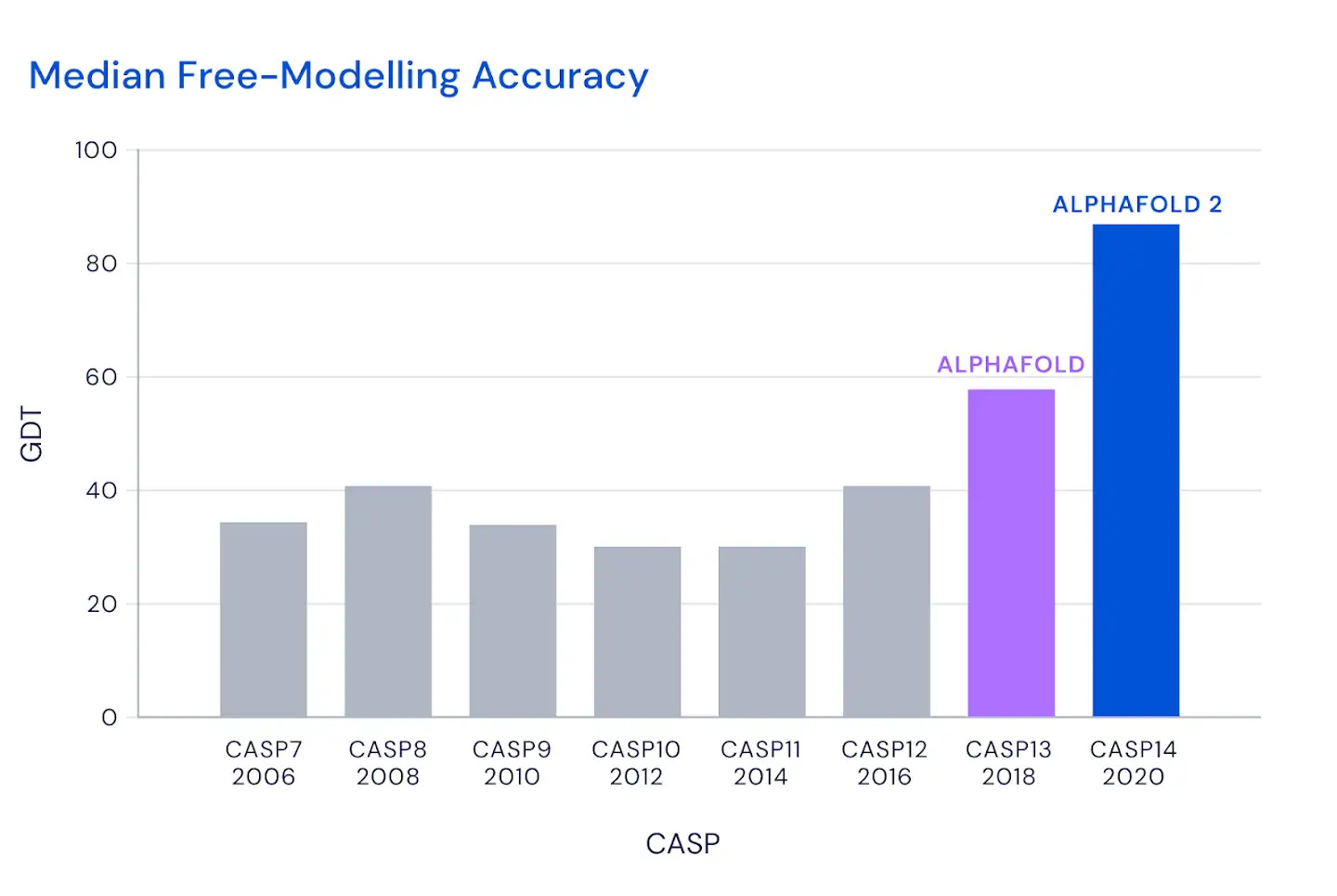
(DeepMind)
AlphaFold si è rivelata meno efficiente nella previsione della forma delle proteine determinata con la risonanza magnetica nucleare, secondo i suoi sviluppatori per via dei dati iniziali diversi da quelli forniti per altre proteine. Il sistema ha inoltre mostrato qualche difficoltà nel prevedere le strutture di singole proteine nei complessi proteici, dove l’interazione tra diverse proteine può portare a una modifica della loro forma.
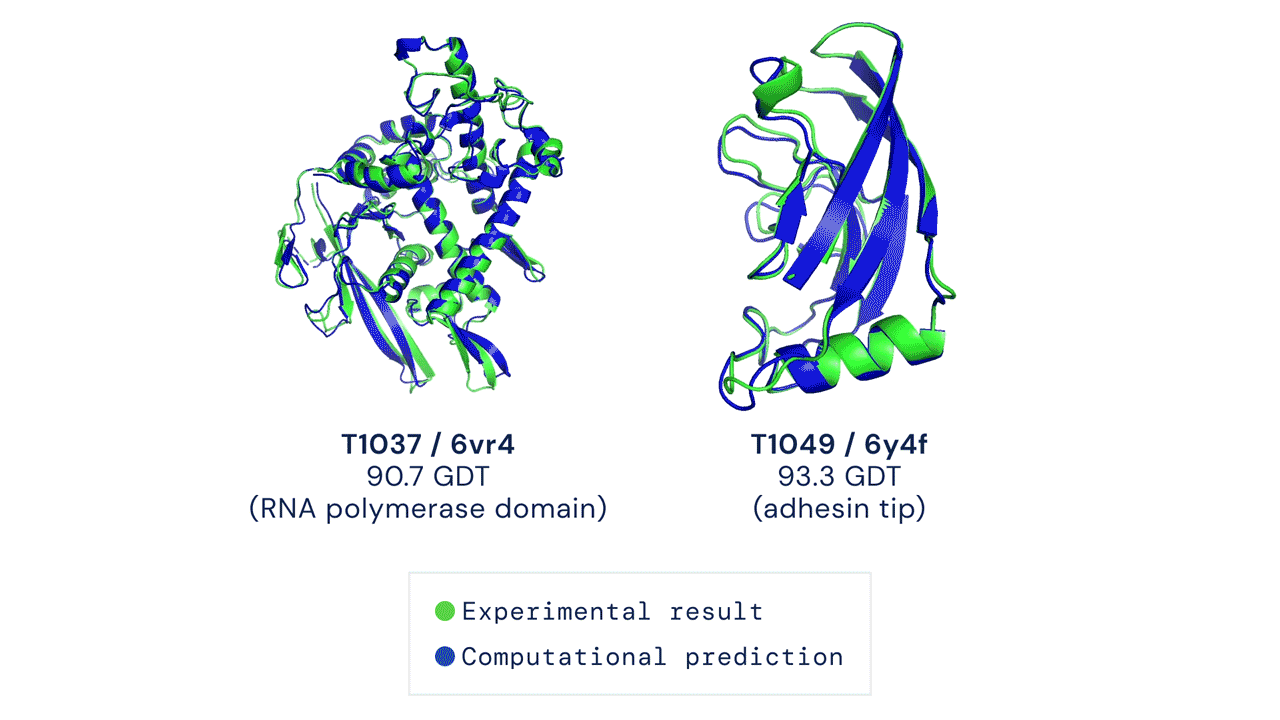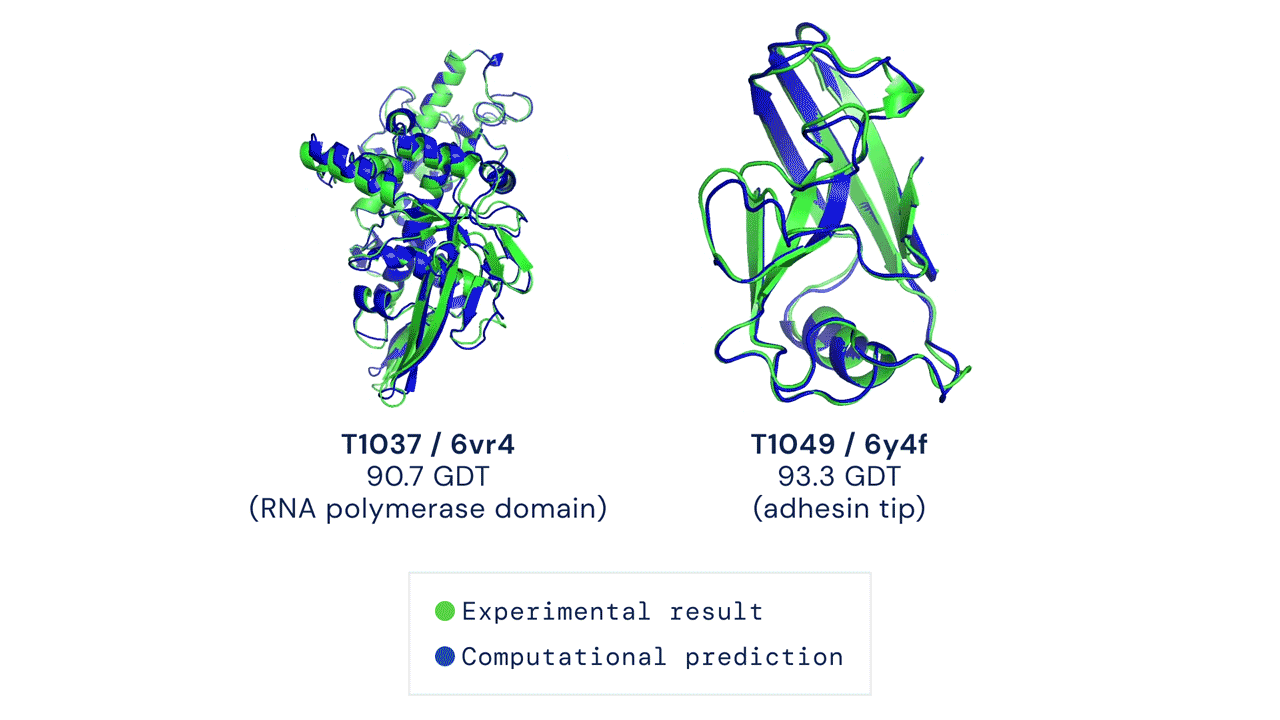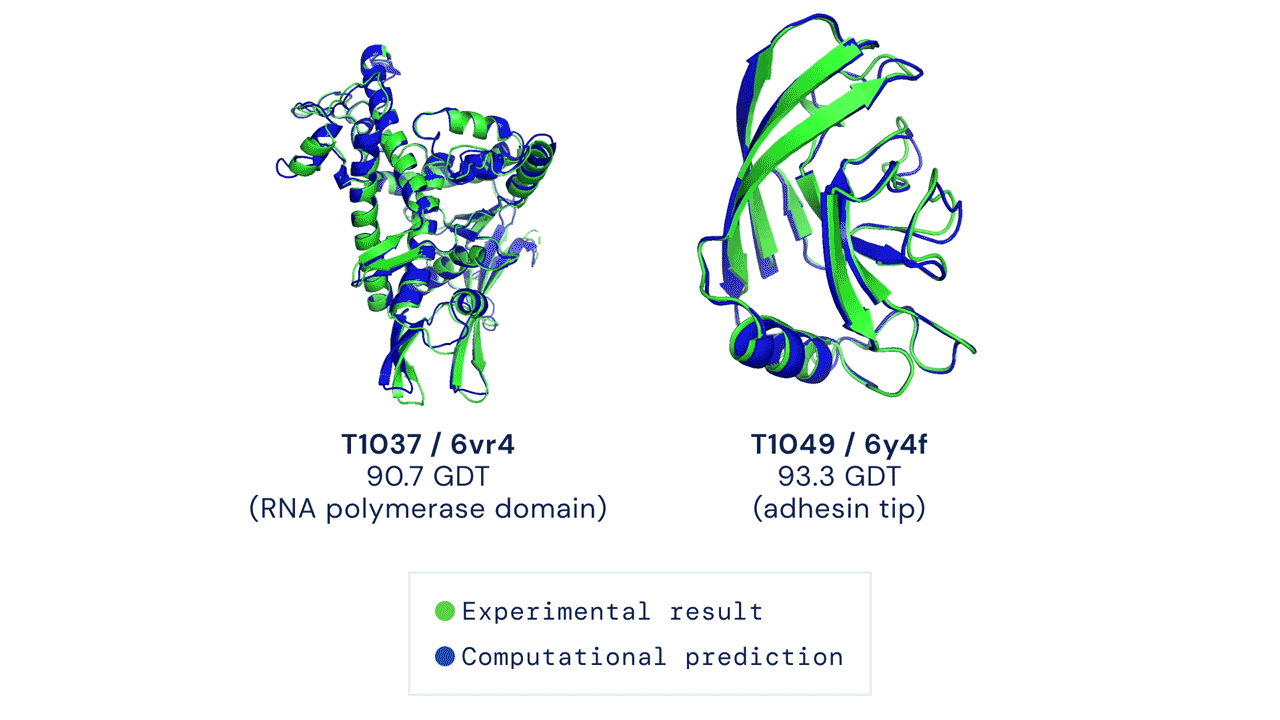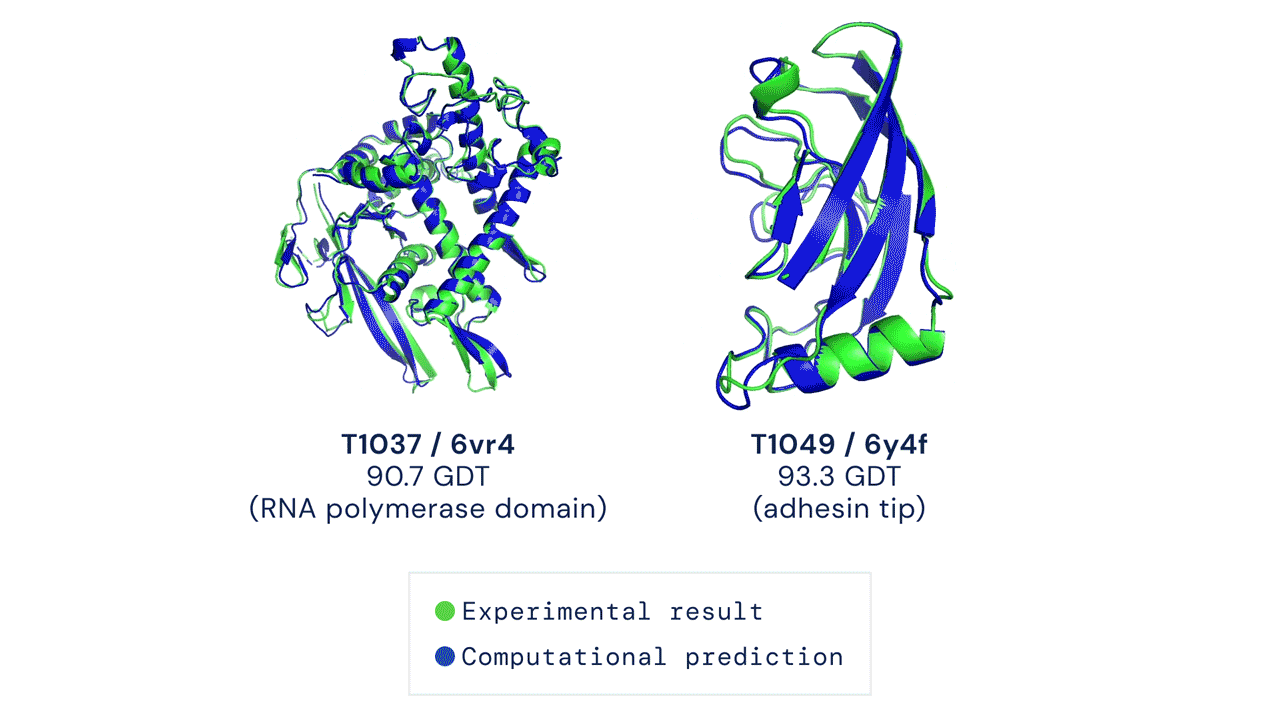
Velocità
Oltre a essere nella maggior parte dei casi molto accurata, l’intelligenza artificiale di DeepMind si è rivelata anche veloce. Ha per esempio impiegato circa mezz’ora per determinare la forma di una proteina batterica sulla quale i ricercatori avevano lavorato per una decina di anni senza ottenere risultati rilevanti. In altre circostanze, il modello ha richiesto alcuni giorni per proporre una soluzione, comprensiva di stime sull’affidabilità della ricostruzione.
Prospettive
I risultati ottenuti da AlphaFold hanno suscitato grande interesse non solo per la loro accuratezza, ma per il salto di qualità rispetto ad appena due anni fa. Il sistema appare molto promettente e se dovesse migliorare ancora potrebbe offrire nuovi importanti strumenti per i ricercatori. Quantità limitate di dati ottenuti in laboratorio potrebbero essere sufficienti per ricostruire la struttura di determinate proteine in poco tempo, accelerandone il loro studio. Il lavoro in laboratorio potrebbe alleggerirsi, consentendo di proseguire con analisi più approfondite sulle proteine oggetto delle ricerche.
Da tempo sappiamo dell’esistenza di migliaia di proteine, sulle quali abbiamo però conoscenze molto limitate proprio perché non siamo ancora riusciti a comprenderne la forma, che determina poi buona parte delle loro funzioni. Un sistema rapido di analisi e di previsione della struttura, come quello che si potrebbe ottenere perfezionando ancora AlphaFold, potrebbe consentire di comprendere meglio alcune caratteristiche del nostro organismo e di alcune malattie, come quelle degenerative e i tumori.